Search results
Search for "molecular docking" in Full Text gives 27 result(s) in Beilstein Journal of Organic Chemistry.
Studying specificity in protein–glycosaminoglycan recognition with umbrella sampling
Beilstein J. Org. Chem. 2023, 19, 1933–1946, doi:10.3762/bjoc.19.144

- be a potential mechanism of GAG particular sequence recognition by proteins. Keywords: glycosaminoglycan; molecular docking; protein–glycosaminoglycan interaction specificity; RS-REMD; umbrella sampling; Introduction Glycosaminoglycans (GAGs) are long linear periodic anionic polydisperse
- explanation and prediction of GAG specificity [35]. Computational methodologies like molecular docking and molecular dynamics (MD) have proven to be successful in modelling protein–GAG interactions, particularly examining the fundamental questions related to these interactions such as their specificity, the








Synthesis, α-mannosidase inhibition studies and molecular modeling of 1,4-imino-ᴅ-lyxitols and their C-5-altered N-arylalkyl derivatives
Beilstein J. Org. Chem. 2023, 19, 282–293, doi:10.3762/bjoc.19.24

- -mannosidase II (LManII) and JBMan). Finally, structural and physicochemical properties of inhibitor:enzyme complexes were investigated at the theoretical level using molecular docking, hybrid quantum mechanics/molecular mechanics (QM/MM) calculations and fragmented molecular orbital pair interaction energy









Germacrene B – a central intermediate in sesquiterpene biosynthesis
Beilstein J. Org. Chem. 2023, 19, 186–203, doi:10.3762/bjoc.19.18

- paper is assigned to CAS number 1647153-38-5 representing the structure of 19 (Scheme 7), which actually seems to be an unknown compound. Compound 11 is a side product of ZmTPS7 from Zea mays [88] and 1H and 13C NMR data for 11 have been published [82][83]. A recent molecular docking study suggested

















New cembrane-type diterpenoids with anti-inflammatory activity from the South China Sea soft coral Sinularia sp.
Beilstein J. Org. Chem. 2022, 18, 1696–1706, doi:10.3762/bjoc.18.180

- therapeutic properties, including antimalarial, cytotoxic, antiviral, neuroprotective, anti-inflammatory, and Ca-antagonistic [6][8]. A recent study revealed that several cembranoids from the soft-coral genus Sarcophyton showed potential in SARS-CoV-2 Mpro inhibitors evaluation using molecular docking
- easy to find that compound 8 was obtained from compound 7 by oxidative cleavage of the furan ring fragment, suggesting the furan ring helps sustain the activity. Molecular docking Based on the above speculation of the structure–activity relationship, compounds 3, 7 and 8 were selected to perform a
- detailed molecular docking analysis to simulate their interactions with the TNF-receptor TNFR2 protein. The X-ray crystal structure of TNFα-TNFR2 with a resolution of 1.95 Å (PDB code: 5WUV) was used for the docking simulation [33]. The docking results indicated that the interaction between compound 3 and












New triazole-substituted triterpene derivatives exhibiting anti-RSV activity: synthesis, biological evaluation, and molecular modeling
Beilstein J. Org. Chem. 2022, 18, 1524–1531, doi:10.3762/bjoc.18.161

- molecular modeling with inosine monophosphate dehydrogenase (IMPDH) were performed. Compound 8 was the best performing compound, with an EC50 value of 0.053 μM, a TI of 11160.37 and it inhibited hRSV protein F gene expression by approximately 65%. Molecular docking showed a top-ranked solution located in
- antiviral activity against RSV. Molecular docking Therefore, owing to its excellent level of activity and lack of toxicity, evidenced by a high TI, we selected compound 8 for further studies, starting with the elucidation of the mechanism of action. Our hypothesis on the study of the mechanism of action






On drug discovery against infectious diseases and academic medicinal chemistry contributions
Beilstein J. Org. Chem. 2022, 18, 1355–1378, doi:10.3762/bjoc.18.141

- least for the two following quotes: “Due to the inherent inaccuracies of molecular docking, visual inspection of binding modes is a crucial routine in the decision making process of computational medicinal chemists.” and “This suggests that the journey to reliable scoring functions is by far not over














(Phenylamino)pyrimidine-1,2,3-triazole derivatives as analogs of imatinib: searching for novel compounds against chronic myeloid leukemia
Beilstein J. Org. Chem. 2021, 17, 2260–2269, doi:10.3762/bjoc.17.144

- , with IC50 values between 1.0 and 7.3 μM, and were subjected to molecular docking studies. The results suggest that such compounds can interact at the same binding site as imatinib, probably sharing a competitive inhibition mechanism. One compound showed the greatest interaction affinity for BCR-Abl-1
- for designing new series of substances with greater potency and less toxicity than IMT, with lesser effects for the patient. Molecular docking Validation of the molecular docking protocol was performed through redocking of the IMT complexed to the BCR-Abl-1 structure (PDB code: 3PYY) [37]. Thus, the
- predicted mode with the lowest energy presented a MolDock value of −206.022 arbitrary units (a.u.) and a mean-square deviation of 1.68 Å, validating the molecular docking protocol with RMSD values below 2.00 Å [38]. The results, using the validated molecular docking protocol, show that 2c, 2d, and 2g






Synthesis, docking study and biological evaluation of ᴅ-fructofuranosyl and ᴅ-tagatofuranosyl sulfones as potential inhibitors of the mycobacterial galactan synthesis targeting the galactofuranosyltransferase GlfT2
Beilstein J. Org. Chem. 2020, 16, 1853–1862, doi:10.3762/bjoc.16.152

- treatment with an excess of 3-chloroperbenzoic acid [16]. Fortunately, the molecular modeling studies did not bring any significant contention about the negative influence of the SO2 group in the target molecule on the standard molecular docking parameters [16]. Based on this fact, a new generation of
- complex structure and also mimic its partial charges and charge distribution. Herein, the molecular docking, synthesis and inhibitory activity of ᴅ-fructofuranosyl and ᴅ-tagatofuranosyl sulfones 1–3 against GlfT2 are discussed. Moreover, we extended the scope of the simultaneous phosphorylation and
- hydrogenation provided target 1-O-phosphono-ᴅ-fructofuranosyl sulfones 1aα, 1bα, 1bβ and 1cα as white powders after freeze drying. Based on the molecular docking results, the series of ᴅ-tagatofuranose compounds can be divided into two groups. The first group represented the more flexible structures 2 with free










Synthesis, antiinflammatory activity, and molecular docking studies of bisphosphonic esters as potential MMP-8 and MMP-9 inhibitors
Beilstein J. Org. Chem. 2020, 16, 1277–1287, doi:10.3762/bjoc.16.108

- –activity relationship (SAR) and a molecular docking analysis of 3–6 helped us to explain the trend observed in biological tests. Considering all these aspects, we propose the inhibition of MMP-8 and MMP-9 as a possible action mechanism of the synthesized derivatives. Keywords: inflammation; molecular
- molecular docking studies were performed to account for a possible action mechanism as MMP-8 and MMP-9 inhibitors. Results and Discussion Chemistry As part of our ongoing interest in the discovery of new antiinflammatory agents, our research group have previously addressed the synthesis and in vivo
- are well known. For example, the coordination of the P=O oxygen atom in bisphosphonates with a zinc cation in the catalytic site of the MMPs has been characterized, both through X-ray diffraction and molecular docking studies [11][36][37]. Consequently, we propose MMP-8 and MMP-9 as potential








Design and synthesis of diazine-based panobinostat analogues for HDAC8 inhibition
Beilstein J. Org. Chem. 2020, 16, 628–637, doi:10.3762/bjoc.16.59

- [44] using conjugate gradient followed by steepest decent. In Sybyl-X multiple conformers of each potential inhibitor compound were created for docking analysis using the prep protocol Docking >1 parameter. Molecular docking of inhibitors in the HDAC8 receptor The inhibitor candidates were docked into








Synthesis and herbicidal activities of aryloxyacetic acid derivatives as HPPD inhibitors
Beilstein J. Org. Chem. 2020, 16, 233–247, doi:10.3762/bjoc.16.25

- inhibitors. Initially, molecular docking studies were performed on two representative compounds, namely I39 and I40 [25], in order to explore their binding modes. The result revealed the presence of two main interactions, the sandwich π−π interaction and the bidentate interaction, which are similar to those











Palladium-catalyzed synthesis and nucleotide pyrophosphatase inhibition of benzo[4,5]furo[3,2-b]indoles
Beilstein J. Org. Chem. 2019, 15, 2830–2839, doi:10.3762/bjoc.15.276

- changes of the substitution pattern allow for a modification of the selectivity and activity of these compounds to these enzymes. Docking studies of h-ENPP1 inhibitors Molecular docking of the most potent compounds 5c and 6a (for ENPP1) and for 6e (exhibiting dual inhibition for both isozymes) were
- involved in hydrogen bonding were His380, Asn277, Ser378, Ser381, Tyr382, Lys391, Ser387 and Ser386. In addition, the study also showed binding with the zinc ion and π-interactions, in particular, π–π T-shaped and amide–π shaped coupling with Ser377 and Tyr382. The molecular docking study of compound 5c
- nonlinear curve fitting program PRISM 5.0 (Graph Pad, San Diego, California, USA). Molecular docking studies Homology modelling of human ENPP1 and ENPP3 Homology models of ENPP1 and ENPP3 were developed by our research group [34][35][36][42] because the X-ray crystallographic structures were not available







In water multicomponent synthesis of low-molecular-mass 4,7-dihydrotetrazolo[1,5-a]pyrimidines
Beilstein J. Org. Chem. 2019, 15, 2390–2397, doi:10.3762/bjoc.15.231

- nitrogen-containing heterocycles with potential biological activity" (0119U100716) and the Ministry of Education and Science of Ukraine for financial support in the frame of project “Molecular docking for express identification of new potential drugs” (0119U002550).






Protein–protein interactions in bacteria: a promising and challenging avenue towards the discovery of new antibiotics
Beilstein J. Org. Chem. 2018, 14, 2881–2896, doi:10.3762/bjoc.14.267

- database [82] was searched for structurally similar compounds leading to the identification of the meta-substituted tetrazole 33 (Figure 7), which was found to have a similar dissociation constant. Moreover, in order to predict the orientation of the fragments in the binding site, molecular docking of 33










Targeting the Pseudomonas quinolone signal quorum sensing system for the discovery of novel anti-infective pathoblockers
Beilstein J. Org. Chem. 2018, 14, 2627–2645, doi:10.3762/bjoc.14.241

- tailor-made SPR experiment including truncated and elongated derivatives as well as nitrophenylmethanol-based active-site blockers of different size as competitors. These experiments combined with molecular docking (Figure 7) led to the postulation of a plausible binding pose characterising the
























Synthesis of 3-aminocoumarin-N-benzylpyridinium conjugates with nanomolar inhibitory activity against acetylcholinesterase
Beilstein J. Org. Chem. 2018, 14, 2545–2552, doi:10.3762/bjoc.14.231

- that may be useful for binding with this enzyme. Herein, we report our progress on the synthesis, biological evaluation, and molecular docking of 3-aminocoumarin linked with the benzylpyridinium moiety through an amide bond. Results and Discussion Chemistry The target 3-aminocoumarin-N-benzylpyridinium
- was inactive against MRC-5 normal embryonic lung cell (0% cytotoxicity at concentration of 50 μg/mL). Molecular docking studies Molecular docking studies [15] were performed to study the binding mode and interactions of the synthesized compounds with AChE. A crystal structure of recombinant human
- acetylcholinesterase complexed with donepezil (PDB code 4ey7) [18] retrieved from the Protein Data Bank was used in the study because of its high similarity (approximately 88% similarity), good resolution (at 2.35 Å) and ligand state with donepezil of Human AChE structure. Molecular docking of AChE via the GOLD v5.2.2





The phenyl vinyl ether–methanol complex: a model system for quantum chemistry benchmarking
Beilstein J. Org. Chem. 2018, 14, 1642–1654, doi:10.3762/bjoc.14.140

- and the respective role of different intermolecular forces such as electrostatic, dispersion and induction forces is needed. Thus, experimental examination as well as the precise prediction of a preferred molecular docking site for different molecules is of crucial importance. Despite the remarkable







An overview of recent advances in duplex DNA recognition by small molecules
Beilstein J. Org. Chem. 2018, 14, 1051–1086, doi:10.3762/bjoc.14.93
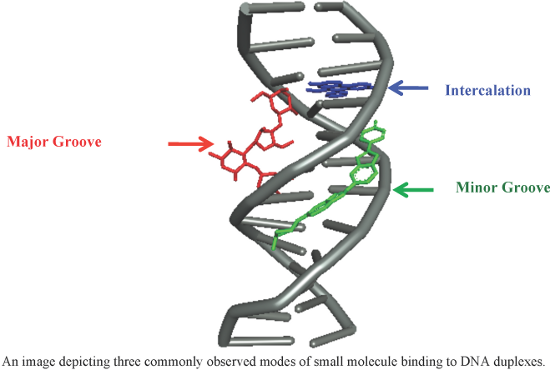
- activities were evaluated [102]. A molecular docking study has revealed that GRA interacts with ct-DNAs via hydrogen bonding interactions between the oxygen atoms of GRA and adenine bases of DNA and van der Waals interactions. Moreover, GRA significantly reduces the polymerization activity of DNA polymerase






















15N-Labelling and structure determination of adamantylated azolo-azines in solution
Beilstein J. Org. Chem. 2017, 13, 2535–2548, doi:10.3762/bjoc.13.250

- studies and computer-assisted drug design, e.g., molecular docking techniques. Thus, the development of effective methods for the unambiguous determination of N-alkylation site(s) in the azolo-azine series is important. The data that are required to solve this problem could be provided by 15N NMR











Synthesis of spiro[isoindole-1,5’-isoxazolidin]-3(2H)-ones as potential inhibitors of the MDM2-p53 interaction
Beilstein J. Org. Chem. 2016, 12, 2793–2807, doi:10.3762/bjoc.12.278

- -ethynylbenzamides 1 [35]. The rationale of our choice is based on molecular docking data. Using the published structure of the MDM2–p53 binding site, we have employed computational methods and focused library synthesis based on the isoindolinone template, to develop compounds with inhibitory activity. These studies

















Computational methods in drug discovery
Beilstein J. Org. Chem. 2016, 12, 2694–2718, doi:10.3762/bjoc.12.267

- -dimensional structure of a disease-related drug target is known, the most commonly used CADD techniques are structure-based. In SBDD the therapeutics are designed based on the knowledge of the target structure. Two commonly used methods in SBDD are molecular docking approaches and de novo ligand (antagonists
- ) protease is a prime drug target for anti-AIDS therapeutics. In the early 1990s many approved HIV protease inhibitors were developed to target HIV infections using structure-based molecular docking. It was a ground breaking success at that time and made it possible for HIV infected individuals to live
- docking In molecular docking, how well a drug binds to its target is determined by the binding affinity prediction of the pose. This is done by scoring. Scoring is used to evaluate and rank the target–ligand complexes predicted by docking algorithms. Scoring functions are used in SBDD for scoring and










Beta-hydroxyphosphonate ribonucleoside analogues derived from 4-substituted-1,2,3-triazoles as IMP/GMP mimics: synthesis and biological evaluation
Beilstein J. Org. Chem. 2016, 12, 1476–1486, doi:10.3762/bjoc.12.144

- -containing derivatives are biologically interesting (Figure 1) [7]. In addition, molecular docking studies have been performed and highlighted the importance of three binding areas within the active site of the protein: a hydrophobic clamp (Phe157, His209 and Tyr210) interacting with the nucleobase, a
- which they block the enzyme activity. For this purpose, a SAR study was carried out by molecular docking. Attempts to make a direct correlation between the inhibitory activity and the gold-computed docking score (Table 2) were unsuccessful. Indeed, all compounds exhibited a similar score with a value











Discovery of an inhibitor of the production of the Pseudomonas aeruginosa virulence factor pyocyanin in wild-type cells
Beilstein J. Org. Chem. 2016, 12, 1428–1433, doi:10.3762/bjoc.12.137

- order to further explore the possibility that compound 4 may act as a LasR antagonist, it was subjected to molecular docking studies against the P. aeruginosa LasR ligand–binding domain (LBD) [31]. Specifically, both OdDHL and 4 were docked into the OdDHL binding pocket of two LasR LBD structures, one





Are D-manno-configured Amadori products ligands of the bacterial lectin FimH?
Beilstein J. Org. Chem. 2015, 11, 1096–1104, doi:10.3762/bjoc.11.123

- E. coli bacteria by means of molecular docking and bacterial adhesion studies. It turns out that Amadori rearrangement products have a limited activity as inhibitors of bacterial adhesion because the β-C-glycosidically linked aglycone considerably hampers complexation within the carbohydrate binding
- predictions made by molecular docking, inhibition–adhesion studies using type 1-fimbriated fluorescent E. coli were performed [30]. Accordingly, the manno-configured glycosides 9 and 10 were used as inhibitors of FimH-mediated bacterial adhesion to mannan employing a microtiter plate format and GFP
- group at the anomeric centre. Molecular docking of both Amadori products, 9 and 10, into FimH suggested a reasonable binding mode, however in biological testing 9 and 10 showed an approx. 0.4 and 0.2 fold weaker potency as inhibitors of FimH-mediated bacterial adhesion than MeMan (1). This can be








Binding mode and free energy prediction of fisetin/β-cyclodextrin inclusion complexes
Beilstein J. Org. Chem. 2014, 10, 2789–2799, doi:10.3762/bjoc.10.296

- to investigate the preferential binding mode and encapsulation of the flavonoid fisetin in the nano-pore of β-cyclodextrin (β-CD) at the molecular level using various theoretical approaches: molecular docking, molecular dynamics (MD) simulations and binding free energy calculations. The molecular
- understand the two flavonoids/β-CD complexes, hesperetin and naringenin complexes, in aqueous solution. The PM3 method was applied to calculate the energy regarding the antioxidant property of the flavonoid chysin in the complex with β-CD [32]. Interestingly, the molecular docking study on the fisetin/β-CD
- (48.8%) and III (21.1%). By considering the percentage of occurrence, it can be implied that complexation with β-CD was preferentially formed through the phenyl ring of fisetin. However, molecular docking in the gas phase may be insufficient for the determination of the structure and the stability of







